Newsletter
> Back to NewsIssue 2021/03
June 2021
Category: Regulatory, FDA
Are you ready to SEND your non-clinical data to FDA?
By Marko Repic
Why? Improved Review of Your Non-clinical Data
Compilation of non-clinical data into a submission dossier is a formidable task for any company preparing to initiate clinical trials or to apply for marketing authorization. No less of a challenge is a review of the documentation by the regulatory agency’s assessors to arrive at the verdict whether the candidate drug is potentially safe to be given to humans. Non-clinical toxicology studies are hundreds of pages long, most of them made up by extensive data tables capturing descriptive statistics of treatment groups and individual animal datapoints for all measured parameters. Therefore, following even a single endpoint in the flood of data tables is not a user-friendly process, as anyone taking a deep dive into the toxicology reports can attest to. To modernize the review of toxicology studies, an initiative by the FDA dating back to 2002, together with Clinical Data Interchange Standards Consortium (CDISC), and Industry has embarked on a journey to harmonize the way the non-clinical study data are organized and presented.
How? SEND Format Allows for Instant Assessment of Data
As an outcome of this process, SEND or Standard for Exchange of Nonclinical Data was implemented defining controlled terminology and format for electronic reporting of non-clinical study endpoints, following a standard data tabulation model developed and curated by CDISC. With the right software, study results in SEND format can be visualized as graphs, charts and tables allowing for instant assessment of multiple study groups, trends, incidences and severities of findings from a single study as well as across multiple studies. Ultimately, SEND improves the quality of data review and can shorten the non-clinical review times on the Agency’s side. On the other hand, utilization of tools to process and visualize SEND data by the industry can bolster the understanding of a drug toxicity profile and guide on mitigation strategies addressing the identified toxicological risks.
Is This Really Needed?
The requirement for submission of SEND data to FDA’s Center for Drug Evaluation and Research (CDER) or FDA’s Center for Biologics Evaluation and Research (CBER) is determined by the study start date, as detailed in Table 1 below.
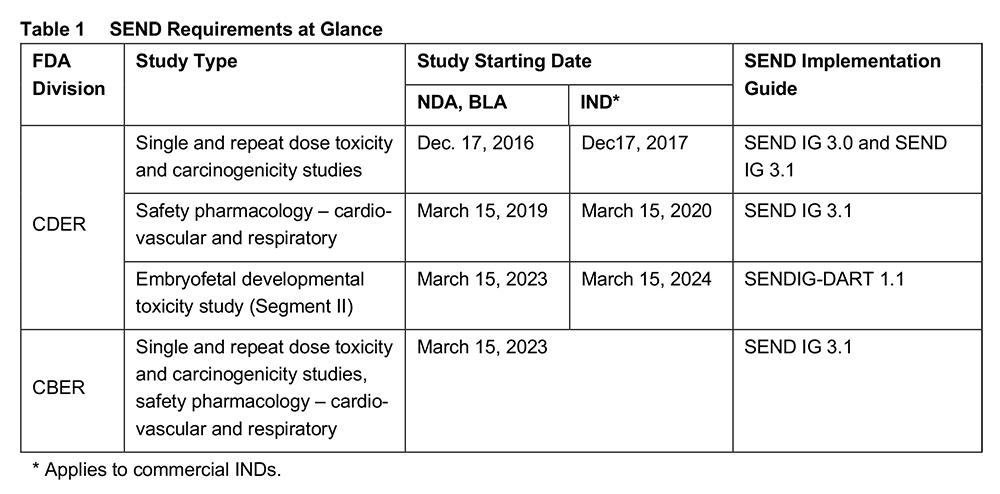
Review of SEND datasets from embryofetal developmental toxicity (Segment II) studies is supported by the FDA from March 15, 2021. SEND data submissions will subsequently become mandatory for studies with a starting date on or after March 15, 2023 (for NDA, BLA) or March 15, 2024 for commercial INDs [2]. It is important to note that the SEND Implementation Guide (IG) for developmental and reproductive toxicity (DART) studies, SENDIG-DART version 1.1, covers Segment II studies only; however, other DART studies will be covered in future versions.
Retrospective generation of SEND data for legacy studies starting prior to the specified dates is not required. However, a simplified trial summary file, specifying the study start date to indicate that that the study is exempt from SEND requirement, needs to be submitted for each legacy toxicology study to avoid technical rejection of the electronic submission [3]. It is also important to note that the GLP status of the non-clinical safety study bears no relevance as SEND is needed even for non-GLP dose range finding studies.
What About Submissions to CBER?
FDA’s Center for Biologics Evaluation and Research (CBER) has also announced its readiness to receive and review SEND data from March 15, 2021 onwards. Submission of SEND data will be mandatory for studies starting on or after March 15, 2023. The affected study types are defined in SEND IG 3.1: single and repeat dose toxicity studies, carcinogenicity studies and safety pharmacology cardiovascular and respiratory studies (Table 1). Support of SEND for DART studies is currently not planned, since standard embryofetal toxicity studies are typically not performed for products reviewed by CBER, however this may change in future. Due to the nature of the products reviewed by CBER, such as vaccines, advanced therapies, safety studies may include specialized endpoints that are not yet covered in SEND. In such cases, electronic submission of nonclinical study results may include data that are modelled in SEND and other endpoints that are not. However, any data that can be modelled as SEND, such as immunogenicity or biodistribution can be helpful to CBER reviewers, as stated in the FDA’s webinar covering this topic [4]. Studies reviewed by the Office of Blood Research and Review (OBRR) are generally not amenable to SEND, and submission requirements should be aligned on with the office on a case-by-case basis.
Take Home Checklist
- Which non-clinical studies in my submission dossier need to be submitted as SEND?
• Check latest SEND implementation guide, FDA Federal Register Notice, or simply talk to us. - Check start dates to distinguish SEND-mandatory vs. legacy studies.
• Don’t forget simplified trial summary file for legacy studies. - Make sure that preparation of SEND files is included in quotes for your upcoming safety studies.
- SEND data not available?
• Get in contact with third party vendors ahead of submission since time to compile these datasets may take several weeks/months.
Happy to Consult you on the Right Path Forward
Concluding, requirements for submission of SEND data are still diverse and not uniform for all types of studies and products. We at MC Tox’Con keep an eye on latest submission requirements in order to ensure your successful regulatory filings.
References
- FDA, Federal register notice. 82 FR 39589. Docket No. FDA-2017-N-2363. 2017.
- FDA, Federal Register Notice. 86 FR 12951. Docket No. FDA-2020-N-2354. . 2021.
- FDA. Technical Rejection Criteria for Study Data. 2021; Available from:
https://www.fda.gov/media/100743/download. - FDA. SEDN for CBER, What You Need to Know. 2020; Available from:
https://www.fda.gov/drugs/news-events-human-drugs/send-cber-what-you-need-know-12042020-12042020.
> Back to News